

Advances in Huntington’s disease therapeutics have triggered renewed interest and investment from drug companies, writes Dr Malvindar Singh-Bains
Huntington’s disease is a debilitating, genetic neurodegenerative disorder, characterised by affective, cognitive, behavioural and motor dysfunctions.1-5
Its aetiology was identified as an abnormally long CAG (cytosine-adenosine-guanosine) trinucleotide repeat expansion in the IT15 gene (Interesting Transcript 15) on chromosome 4, which encodes a mutant protein called huntingtin.6
Worldwide, Huntington’s disease has a prevalence of five to 10 per 100,000 in Australia, South America, North America, and most European countries, but a lower prevalence of 0.5:100,000 in Asia, and even lower in Africa.3
Interestingly, the region with the highest prevalence of Huntington’s disease in Australia is Tasmania, with 12.1 cases per 100,000 people.7
Depending on the individual CAG expansion, Huntington’s disease symptom onset typically ranges from 35 to 44 years of age. However, special cases have been reported for ages as young as four and as old as 80.8, 9
Typically, after the onset of symptoms, the lethal progression and death from the disease is 10 to 20 years.10
Despite the aetiology of Huntington’s disease as a single-gene disorder, there is considerable variability in the types of motor, behavioural and cognitive symptoms both at clinical outset and thereafter during disease progression.11, 12
MOTOR DYSFUNCTION
Typically, the onset of motor symptoms is used to define the clinical onset of the disease.6,11 The motor abnormalities associated with Huntington’s disease affect both involuntary and voluntary actions.
Development of an involuntary “choreoathetotic” disorder, which involves rapid, irregular, involuntary choreiform (Latin for “dance-like”) movements, are one of the most distinct and well recognised symptoms of the disease, which was documented in George Huntington’s original description of the disease
in 1872.13
Chorea is characterised by sudden, quick, unintended movement of almost any part of the body, including the face and limbs.14 Although the presence of chorea is useful for the diagnosis of the disease, it is a poor marker of disease severity.
Analyses of Huntington’s disease populations have shown that approximately 50 to 70% of cases present with chorea at onset.15, 16
The hyperkinetic syndrome is progressively replaced by a more hypokinetic (akinetic-rigid) syndrome which involves bradykinesia, rigidity and dystonia.
Furthermore, the patient’s ability to speak and swallow is also affected, which can leave the patient susceptible to aspiration pneumonia secondary to dysphagia, and is the most common cause of death in Huntington’s disease.3, 11, 14, 17-19 ‘
COGNITIVE IMPAIRMENT
In terms of cognitive impairment, the symptoms begin early and become more severe as the disease progresses.
Deficits in cognition tend to begin with the loss of mental flexibility and a gradual decline of intellectual processes that can lead to profound dementia.
Early cognitive deficits include difficulties in concentration, dysfunction in short-term memory, and executive function impairment.20-22
As disease progression continues, these cognitive deficits can advance into widespread dementia, in addition to verbal language deterioration and difficulties in visuospatial function.20, 23, 24
BEHAVIOURAL AND PSYCHIATRIC CHANGES
Behavioural and psychiatric symptoms in Huntington’s disease are common, with 30 to 50% of symptom presentation at disease onset being behavioural and emotional problems such as irritability, aggression, anxiety, and obsessive behaviour, and most commonly, depression.15,16 There is a vast range of mood and psychiatric symptoms including depression, dysphoria, agitation, irritability, labile mood, apathy and anxiety.25, 26 Other symptoms can include obsessive-compulsive behaviours, sleep disturbances, personality changes, psychosis, and suicidal ideation, with suicide being estimated in patients with the disease to be about five to 10 times that of the general population.3, 27-30
DIAGNOSIS
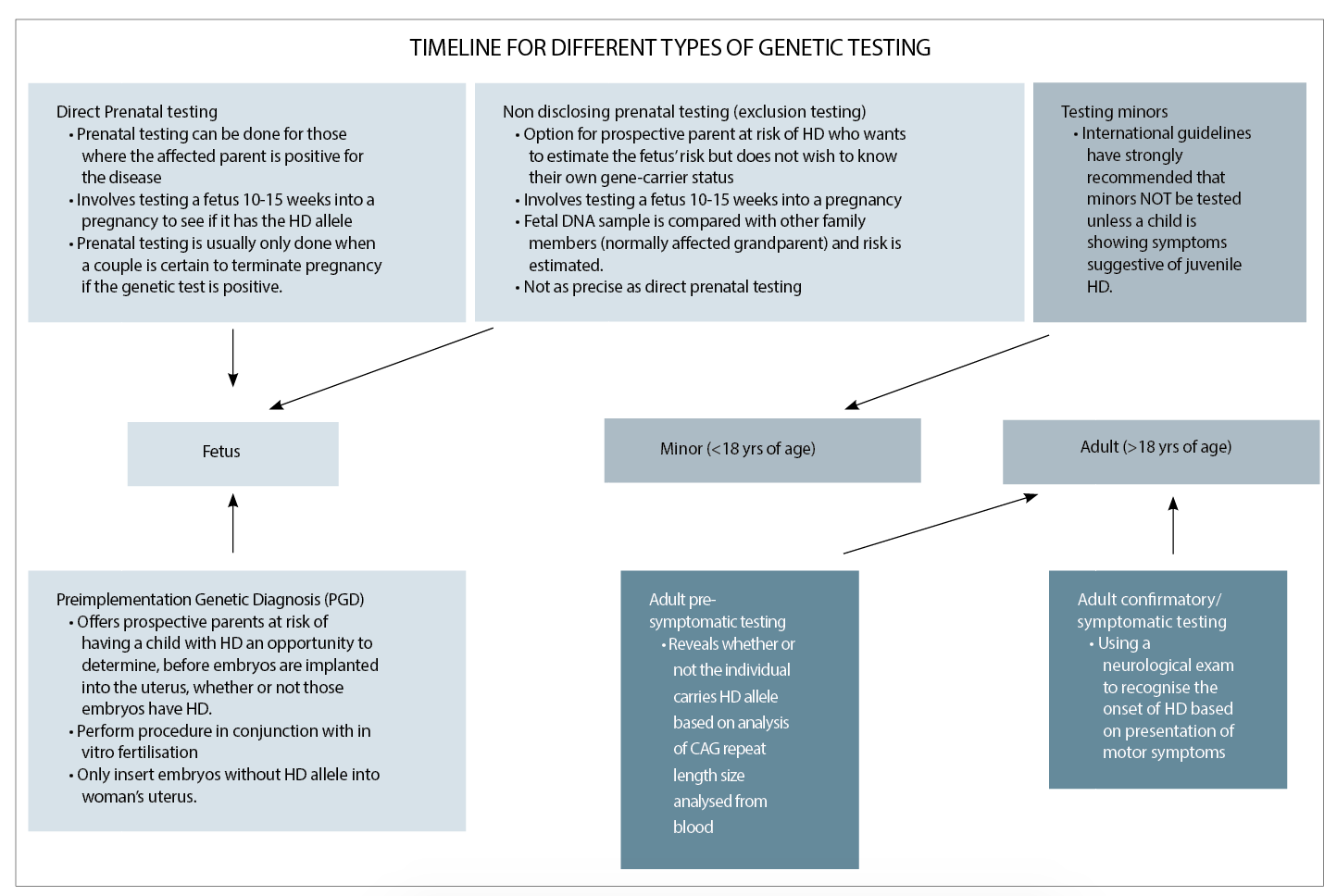
In 1993, the Huntington’s Disease Collaborative Research Group isolated the Huntington gene and identified the hereditary version of this gene that causes the disease.6
They discovered that in all people, the three-letter codon sequence C-A-G is repeated several times at one end of the Huntington gene. The discovery of the genetic variant that is responsible for the disease allowed for the development of a highly accurate direct gene test for it. (See table above)
Huntington’s disease is inherited in an autosomal dominant pattern, meaning that inheriting only one copy of the disease allele from a parent is sufficient to cause an individual to develop the disease.31 Thus, a child who has one affected parent has a 50% risk of inheriting the mutated version of the Huntington gene and eventually developing the disease.
This “50% risk” reflects the equal probabilities that either the individual carries the disease allele and will develop Huntington’s disease; or, that the individual has all non-disease alleles and will not develop the condition. The genetic nature of Huntington’s disease implies that there must be a family history of the condition in order for one to be at risk for developing the disorder.
However, there are exceptions. That is, if a family member with the disease died at an early age before symptom onset, or they were misdiagnosed with another disorder with Huntington’s disease-like symptoms; or, in very rare cases, a new mutation was documented.
Normal individuals tend to have CAG repeats of 36 or less, and will not develop Huntington’s disease. However, incomplete penetrance of the phenotype can occur at 36-39 repeats, which means it is uncertain if the individual will develop symptoms during life and is considered the “grey area”.
TREATMENT
While no existing drugs can actually stop or reverse the neurological degeneration that Huntington’s disease causes, most if not all patients can benefit from the management of certain symptoms associated with the condition, such as chorea, psychosis, and depression.
Treatment of chorea
Tetrabenazine (Xenazine) was granted FDA approval in 2008 as a drug to provide relief from chorea in Huntington’s disease. Other drugs are also used to help reduce involuntary movements – these include neuroleptics and benzodiazepines. Typical neuroleptics such as haloperidol or fluphenazine are quite effective. Some atypical neuroleptics such as olanzapine and risperidone may also be effective.
Treatment of dystonia
Pharmacologic treatment of dystonia in Huntington’s disease may include benzodiazepines, baclofen, and sometimes dopaminergic agents developed for Parkinson’s disease. Botulinum toxin injections can be quite effective for focal dystonias.
Psychiatric treatment
Pharmacologic treatment of psychiatric symptoms in Huntington’s disease often involve the use of neuroleptics including atypical antipsychotics such as Olanzepine, Ziprasidone, Quetiapine and Aripiprazole which are used for psychosis and sometimes for irritability or chorea suppression.
High potency antipsychotics can also be used such as haloperidol, pisperidone, or fluphenazine (sometimes also used to treat chorea). Antidepressents such as SSRIs can be used for depression and sometimes for anxiety. Benzodiazepines may be a good choice for short-term management of agitation.
RESEARCH
Since the discovery of the Huntington’s disease gene in 1993, researchers believed a cure would soon follow as the gene would be a clear therapeutic treatment target. However, researchers quickly realised that the protein product of the gene, huntingtin, is essential for embryonic development and is involved in diverse cellular activities such as vesicular transport, endocytosis, autophagy and the regulation of transcription.35
Furthermore, the large 348kda protein has been extremely difficult to structurally characterise due to its vast complexity – posing a challenge for drug development.
Nevertheless, researchers are covering huge ground and hope is on the horizon. With the help of cryo-electron microscopy researchers from the Max Planck Institute of Biochemistry in Martinsried and Ulm University have now decoded the three-dimensional, molecular structure of the healthy human huntingtin protein, paving the way for functional studies.36
Knowing the structure of human huntingtin will enable the identification of potential binding sites for future therapeutic agents.
Recent advances in genome editing and gene-silencing technology have opened up a whole new field for the treatment of genetic conditions such as Huntington’s disease.
Some gene-silencing techniques, such as antisense oligonucleotides, have been around for decades. Newer techniques, notably genome editing tools such as CRISPR/Cas9, have only been discovered and developed over the last few years.
The most exciting development in disease therapeutics to date is the recent announcement by Ionis and Roche that the first human trial of a huntingtin-lowering drug, antisense oligonucleotide IONIS-httrx, demonstrates that it reduces mutant huntingtin by up to 60% in the cerebral spinal fluid, and is safe and well-tolerated.
This phase 1/2 clinical trial involving 46 patients with early-stage disease were treated for 13 weeks with four intrathecal injections of 10mg, 30mg, 60mg, 90mg or 120mg of IONIS-httrx (RG6042) or placebo, administered monthly. An open-label extension (OLE) study for patients who participated in the Phase 1/2 study is ongoing.
The advances in Huntington’s disease therapeutics have triggered renewed interest and investment from drug companies, while researchers continue to refine bigger and better animal models of Huntington’s disease for preclinical testing.
Scientists continue to discover new clinicopathological features of the disease through the study of post-mortem human tissue in New Zealand and around the world.37, 38
This is the most exciting era for the Huntington’s disease community – for clinicians, scientists and, most importantly, for the families affected by the disease.
Dr Malvindar Singh-Bains, PhD, is a Freemasons Research Fellow at the Centre for Brain Research at the University of Auckland and co-chair of Huntington’s Disease Youth Organisation NZ
References:
1. Albin RL, Tagle DA. Genetics and molecular biology of Huntington’s disease. Trends Neurosci. 1995;18(1):11-4.
2. Wilson RS, Como PG, Garronl DC, Klawans HL, Barr A, Klawans D. Memory failure in Huntington’s disease. J Clin Exp Neuropsychol. 1987;9(2):147-54.
3. Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218-28.
4. Nance MA. Huntington disease: clinical, genetic, and social aspects. J Geriatr Psychiatry Neurol. 1998;11(2):61-70.
5. Margolis RL, Ross CA. Diagnosis of Huntington disease. Clin Chem. 2003;49(10):1726-32.
6. MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971-83.
7. Pridmore SA. The prevalence of Huntington’s disease in Tasmania. The Medical journal of Australia. 1990 Aug 6;153(3):133-4.
8. Harper P. In Huntington’s Disease: W. B. Saunders; 1991.
9. Haslam R, Curry B, Johns R. Infantile Huntington’s Disease. Can J Neurol Sci. 1983;10(3):200-3.
10. Reiner A, Dragatsis I, Dietrich P. Genetics and neuropathology of Huntington’s disease. Int Rev Neurobiol. 2011;98:325-72.
11. Waldvogel HJ, Kim EH, Thu DC, Tippett LJ, Faull RL. New Perspectives on the Neuropathology in Huntington’s Disease in the Human Brain and its Relation to Symptom Variation. J Huntington’s Disease. 2012;1(2):143-53.
12. Claes S, Van Zand K, Legius E, Dom R, Malfroid M, Baro F, et al. Correlations between triplet repeat expansion and clinical features in Huntington’s disease. Arch Neurol. 1995;52(8):749.
13. Huntington G. On chorea. Med Surg Rep. 1872;26:317-21.
14. Folstein SE. Huntington’s Disease: a disorder of families. Baltimore: Johns Hopkins University Press; 1989.
15. Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Onset symptoms in 510 patients with Huntington’s disease. J Med Genet. 1993;30(4):289-92.
16. Witjes-Ané MW, Zwinderman A, Tibben A, van Ommen GB, Roos R. Behavioural complaints in participants who underwent predictive testing for Huntington’s disease. J Med Genet. 2002;39(11):857-62.
17. Young AB, Shoulson I, Penney JB, Starosta-Rubinstein S, Gomez F, Travers H, et al. Huntington’s disease in Venezuela Neurologic features and functional decline. Neurology. 1986;36(2):244-.
18. Berardelli A, Noth J, Thompson PD, Bollen EL, Currà A, Deuschl G, et al. Pathophysiology of chorea and bradykinesia in Huntington’s disease. Mov Disord. 1999;14(3):398-403.
19. Mahant N, McCusker E, Byth K, Graham S. Huntington’s disease Clinical correlates of disability and progression. Neurology. 2003;61(8):1085-92.
20. Montoya A, Price BH, Menear M, Lepage M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J Psychiatry Neurosci. 2006;31(1):21.
21. Snowden J, Craufurd D, Thompson J, Neary D. Psychomotor, executive, and memory function in preclinical Huntington’s disease. J Clin Exp Neuropsychol. 2002;24(2):133-45.
22. Ho A, Sahakian B, Brown R, Barker R, Hodges J, Ané M-N, et al. Profile of cognitive progression in early Huntington’s disease. Neurology. 2003;61(12):1702-6.
23. Kirkwood SC, Su JL, Conneally PM, Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol. 2001;58(2):273.
24. Lawrence AD, Watkins LH, Sahakian BJ, Hodges JR, Robbins TW. Visual object and visuospatial cognition in Huntington’s disease: implications for information processing in corticostriatal circuits. Brain. 2000;123(7):1349-64.
25. Rosenblatt A, Leroi I. Neuropsychiatry of Huntington’s Disease and other basal ganglia disorders. Psychomatics. 2000;41:24-30.
26. Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-4.
27. Beglinger LJ, Langbehn DR, Duff K, Stierman L, Black DW, Nehl C, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-8.
28. Anderson KE, Louis ED, Stern Y, Marder KS. Cognitive correlates of obsessive and compulsive symptoms in Huntington’s disease. Am J Psychiatry. 2001;158(5):799-801.
29. Baliko L, Csala B, Czopf J. Suicide in Hungarian Huntington’s disease patients. Neuroepidemiology. 2004;23(5):258-60.
30. Robins Wahlin TB, Bäckman L, Lundin A, Haegermark A, Winblad B, Anvret M. High suicidal ideation in persons testing for Huntington’s disease. Acta Neurologica Scandinavica. 2000;102(3):150-61.
31. Myers R, MacDonald M, Koroshetz W, Duyao M, Ambrose C, Taylor S, et al. De novo expansion of a (CAG) n repeat in sporadic Huntington’s disease. Nat Genet. 1993;5(2):168-73.
32. Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369-84.
33. Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman J-J, et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet. 1996;59(1):16.
34. McNeil SM, Novelletto A, Srinidhi J, Barnes G, Kornbluth I, Altherr MR, et al. Reduced penetrance of the Huntington’s disease mutation. Hum Mol Genet. 1997;6(5):775-9.
35. Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016 Mar 2;89(5):910-26.
36. Guo Q, Bin H, Cheng J, Seefelder M, Engler T, Pfeifer G, et al. The cryo-electron microscopy structure of huntingtin. Nature. 2018;555:117.
37. Singh Bains MK, Tippett LJ, Hogg VM, Synek BJ, Roxburgh RH, Waldvogel HJ, et al. Globus pallidus degeneration and clinicopathological features of huntington disease. Annals of neurology. 2016;80(2):185-201.
38. Mehrabi NF, Waldvogel HJ, Tippett LJ, Hogg VM, Synek BJ, Faull RLM. Symptom heterogeneity in Huntington’s disease correlates with neuronal degeneration in the cerebral cortex. Neurobiol Dis. 2016;96:67-74.
