

Given the cost of developing new therapeutic drugs, should we not repurpose existing drugs to better address neurological diseases and conditions?
The experience of many patients diagnosed with malignant brain cancer is often similar. Pre-diagnosis experiences tell of “not feeling quite right” for a few weeks or months, persistent headaches and periodic numbness of the extremities.
What then triggers a visit to the doctor and the diagnosis, is usually, the onset of otherwise unexplained seizures.
A CT scan, or other imaging test, which indicates some type of abnormal growth in the brain confirms the unexpected and confronting diagnosis and prognosis.
The neuroanatomical location of the tumour will likely explain the specific symptoms experienced by each patient. It is important to note that the above-mentioned symptoms do not mean that a tumour is the problem; the clinical tests provide the evidence and confirm the underlying symptom trigger.
Brain cancer constitutes about 2% of cancers overall, but it is a high impact disease.
In Australia, there were 1596 new cases (2009) and 1247 deaths (2010) from malignant brain cancers. In 2012, brain cancer was estimated to account for 21,500 disability adjusted life years (DALYs) in Australia; of these, 20,200 were years lost due to premature death and 1300 were years of healthy life lost due to the burden of disease.
Brain cancer is the leading cause of cancer death in children aged under 15 and adults aged to 39 years, and the second leading cause of cancer death in females aged 0-44, second only to breast cancer.1
Aside from age, no risk factors have been identified and no screening procedures are in place. As with many cancers, if the proportion of the elderly in the population increases, the incidence of brain tumours is expected to rise.
It is important to consider that brain cancer is a complex and diverse array of diseases affecting the brain and spinal cord. In this article, I focus on brain cancers which are not the result of metastasis from another site, outside the central nervous system (CNS), but primary brain cancers which begin in and affect the CNS.
Interestingly, primary brain cancers, unlike most other cancers, rarely metastasise to organs outside the CNS, which means that tumour cells from lung, breast, skin cancer can enter the brain but not the other way around. Why this is so, is presently unclear, but is likely due to the differential synthesis and/or activation of molecules involved in migration and invasion.
Brain cancers are distinguished, based on the regions of the brain affected and the types of cells involved. Further classification is based on the age of patients, defining the so-called paediatric (e.g. medulloblastoma) and adult brain cancers (e.g. glioma, meningioma). Here, I will focus on the most common non-paediatric brain tumour, which is also the most common and aggressive type, glioblastoma (GBM).
Sadly, 75% of GBM patients will die within five years of diagnosis. Even with the incredible advances in surgery and radiotherapy, the prognosis for patients has remained grim for decades and the complications associated with the toxicity of limited standard chemotherapy treatments severely reduce quality of life of many patients.
One of the most significant advances in therapy came, not from a drug discovery breakthrough, but a reconfiguration of existing therapies involving carefully timed surgery, radiotherapy in combination with chemotherapy. This is the so-called “Stupp protocol” developed by Roger Stupp and his Swiss colleagues in 2005.2
Stupp and colleagues studied almost 600 patients with malignant brain cancer and followed their progress following various treatment strategies over several years. Their new therapeutic protocol demonstrated a 2.5-fold increase in the survival rate, from 10% to 27%, at two years’ post-diagnosis, compared with the pre-Stupp protocol.
The current standard drug used is temozolomide (TMZ); a DNA alkylating agent which kills dividing tumour cells but the broad non-specific cytotoxic mechanism of drugs such as TMZ is also responsible for negative side-effects.
Despite the limited advances in therapy, the last 10 years has seen a massive increase in understanding the molecular and genetic underpinnings of brain cancer, especially GBM. In a coordinated international effort, hundreds of GBM biopsies have been analysed, backed by state-of-the-art DNA and RNA sequencing technology.3
These genomic studies have revealed discrete molecular signatures and mutations which have further allowed sub-classification of GBM into at least four distinct molecular groups.
Why is this important? Because better understanding of the mechanisms driving carcinogenesis will facilitate the discovery of more specific drug targets and diagnostic biomarkers, which in turn will lead to more effective patient-specific therapies.
Developing a new therapeutic drug is a multi-stage process that involves identifying and validating a molecular target and then developing drugs. This is an expensive process costing more than $US1 billion for every successful FDA drug registration.
The economics of this mean that major drug companies (“big pharma”) understandably prefer to target diseases with high economic impact. Since brain cancers represent only about 2% of all cancers diagnosed, big pharma has tended to not invest in new brain cancer drug development.
One way of bypassing the enormous cost of drug development with diseases that attract less attention from major pharmaceutical companies is to repurpose existing drugs. Many drugs have a wide range of actions that benefit many diseases, particularly in combination with existing therapies.
The first step in this approach is to identify a molecular target important for promoting a disease state.
We recently discovered an unusual molecular mechanism involved in brain cancer cell suicide (apoptosis). It turns out that activation of cAMP (cyclic-AMP) in three of the six GBM cell lines tested (each cell line originated from a different patient) caused rapid apoptosis.4
I’m sure if you think back to your textbook biochemistry, you will recall that the cAMP pathway was the first molecular pathway described; a pathway typically involved in the activation of many cell functions following hormone or neurotransmitter stimulation.5
As mentioned, three of six GBM cells were killed by acute cAMP activation. However, the three other GBM cells appeared to be resistant at the drug doses tested. We found that the three resistant cells showed elevated activation of another cell signalling pathway, the MAPK pathway, which protected the cells.
Luckily, the MAPK pathway is a well-studied pathway in cancer cells and there are many experimental drugs blocking MAPK signalling available to researchers. When we inhibited the MAPK pathway and activated cAMP at the same time, these formerly resistant cells, were killed.
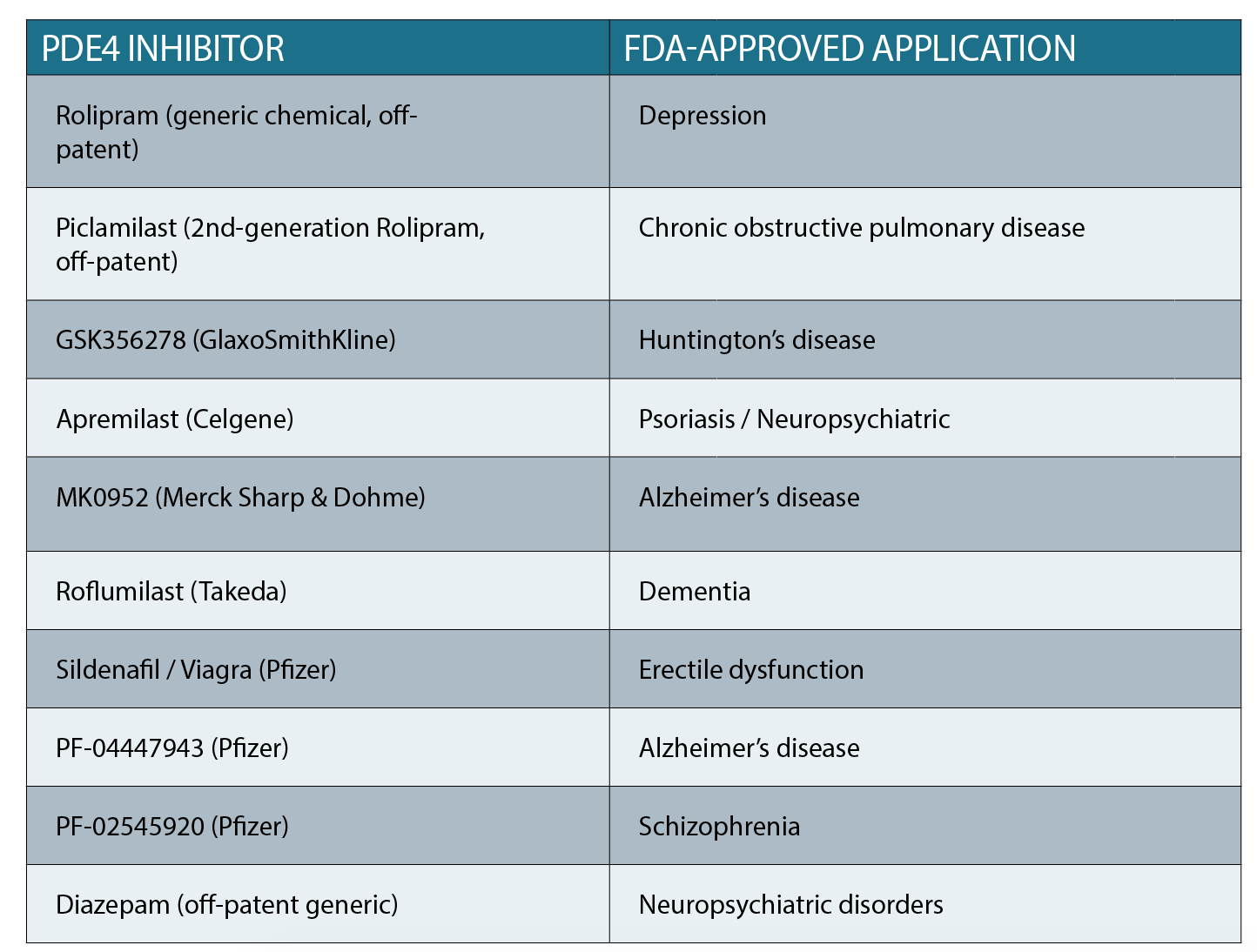
So how do we use this new information to advance GBM therapy? Our hope is that we can use existing FDA-approved drugs which are used for various conditions, including neurological diseases/conditions (See table above).
These drugs inhibit a group of enzymes called phospho-diesterases (PDEs). Importantly, PDEs are natural inhibitors of the cAMP pathway. So, inhibiting the inhibitor (the PDE) will stimulate cAMP activation, which in turn can trigger GBM cell death.
Depending on the patient’s cells, PDE inhibitors and standard therapy would be combined with other drugs, including MAPK inhibitors.
Crucially, since many of the FDA-approved PDE inhibitors are targeted at brain diseases, there is proven brain bioavailability – a shortcoming of many other drugs proposed to be repurposed for brain cancer.
Can we convince clinicians to add one of the PDE inhibitor drugs to a GBM patient’s therapeutic protocol? Given that we propose testing already FDA-approved drugs, perhaps?
Of course, there will be side-effects which are known in non-cancer patients, but will there be severe side-effects in a GBM patient who has undergone, or is undergoing, surgery radiotherapy and chemotherapy? Another question is, which of the many available PDE inhibitors should be tested?
One avenue for proof-of-concept pre-clinical research, is to use a non-human surrogate organism to conduct tests using novel experimental therapies.
Our best option currently, is a mouse model of brain cancer. We have developed a brain-cancer mouse model in which we can, at will, trigger specific mutations which begin a train of cellular transformation events leading to brain cancer which resembles GBM.6 These will be the “patients” that may reveal whether PDE inhibitors, in combination with standard TMZ chemotherapy, can significantly slow tumour growth.
Ideally, we would also like to combine radiotherapy in the pre-clinical experiment to mimic the human therapy as closely as possible.
Ultimately, time will tell but the acceleration in the body of knowledge, both clinical and biological, gives us confidence that therapy for malignant brain cancers will improve, sooner rather than later.
Theo Mantamadiotis, PhD, is a senior lecturer and laboratory head for the Department of Microbiology & Immunology and Department of Surgery (RMH), The University of Melbourne
References:
1. Cancer in Australia: an overview, 2012 Cat. no. CAN 70. Canberra: AIHW ed: Australian Institute of Health and Welfare & Australasian Association of Cancer Registries 2012; 2012.
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987-96.
3. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-77.
4. Daniel PM, Filiz G, Mantamadiotis T. Sensitivity of GBM cells to cAMP agonist-mediated apoptosis correlates with CD44 expression and agonist resistance with MAPK signaling. Cell Death Dis. 2016;7(12):e2494.
5. Sassone-Corsi P. The cyclic AMP pathway. Cold Spring Harb Perspect Biol. 2012;4(12).
6. Daniel P, Filiz G, Brown D, Christie M, Waring P, Zhang Y, et al. PI3K Activation In Neural Stem Cells Drives Tumorigenesis Which Can Be Suppressed By Targeting CREB. BioRxiv. .
